Research
Precision Genomics in Neurodevelopmental Disorders
PRECISION GENOMICS:
Inspired by the term Precision Medicine, we defined Precision Genomics as “determining all possible relevant variation within an individual to the precise nucleotide.” Precision Genomics is not equivalent to Precision Medicine but is a key aspect under the umbrella of Precision Medicine similar to other areas (e.g., Precision Psychiatry, Precision Imaging, etc.). As a research lab, we excel to achieve the goals of Precision Genomics by addressing five main areas that present as current limitations to Precision Genomics in everyone:
- interpretation of noncoding variation
- variants missed due to genomic technology
- speed of the current “genomic workflow”
- combination of multi-hit rare and/or common variants
- gene x environment
Currently, the main focus of our lab is to pursue each of these in neurodevelopmental disorders. Each area is outlined below.
1. INTERPRETATION OF NONCODING VARIATION:
Neurodevelopmental disorders (NDDs) affect >1% of the population. Considerable progress has been made in understanding the contribution of rare protein-coding variants and large copy number variants in NDDs. However, the role of rare noncoding variation has been less clear due to the limited number of individuals with whole-genome sequencing (WGS) data previously available. We were one of the first to identify aggregate enrichment for promoters and enhancers in the first 516 families assessed by WGS; signals that have now been seen by multiple additional groups. In our lab, we are working on the interpretation of noncoding variation by analyzing whole-genome sequencing data from additional families, developing statistical strategies to assess noncoding variation in this data, and performing massively parallel reporter assays to assay quantitative in vitro effects of variation in these regions followed by detailed characterization of top noncoding regions.
The first publication from our lab on noncoding variation in a specific noncoding region in neurodevelopmental disorders was “Coding and noncoding variants in EBF3 are involved in HADDS and simplex autism.” ln this paper, we identified an excess of de novo variants in an enhancer element called hs737. We found that hs737 targets the gene EBF3; a gene genome-wide significant for protein-coding de novo variation in NDDs. By combining multiple data types (i.e., genomic, phenotypic, functional) we proposed a gene regulatory network involving hs737 and its target gene EBF3. Check out the gallery to see figures, from our paper, related to this work.
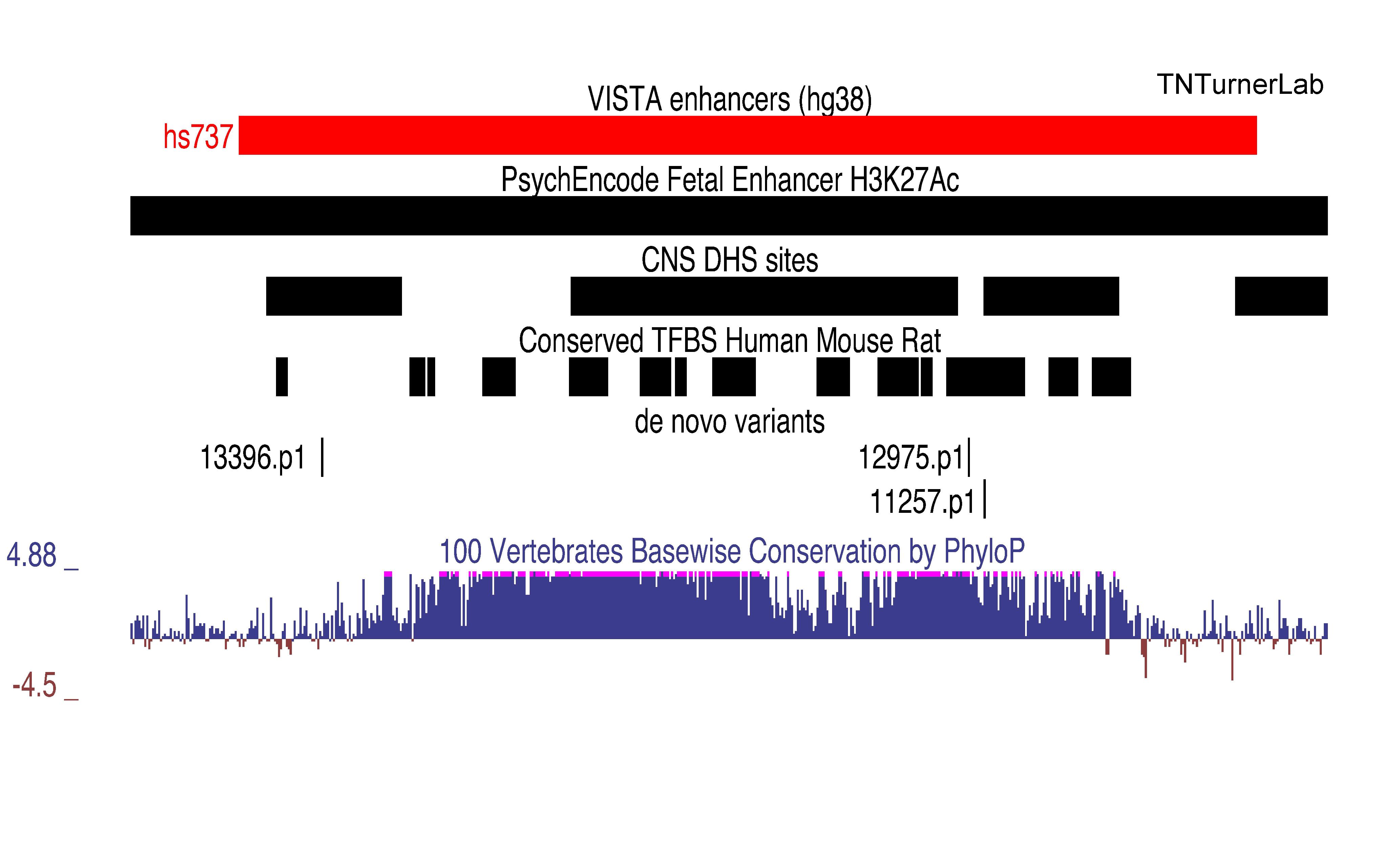
hs737 enhancer
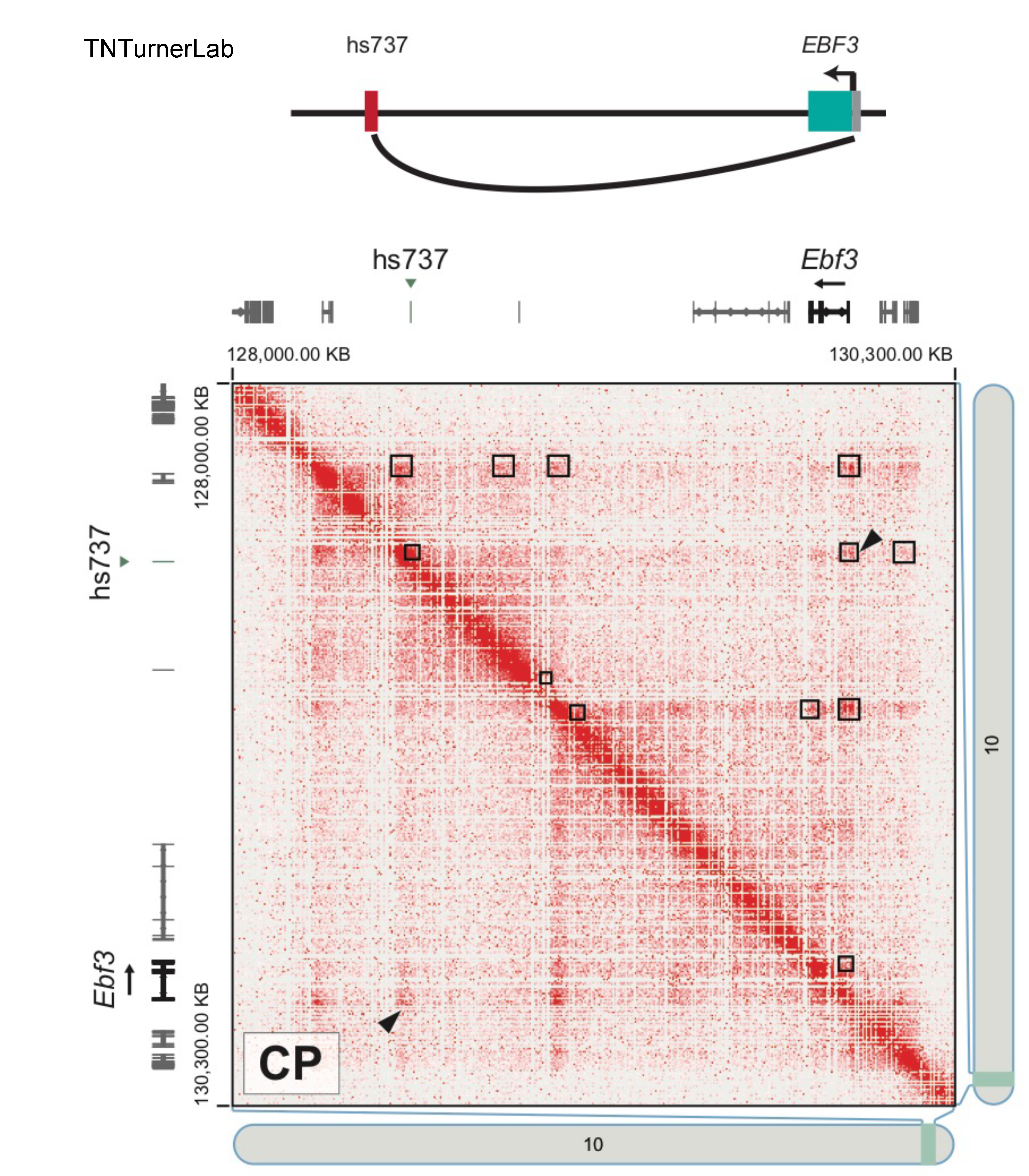
The target gene of hs737 is EBF3
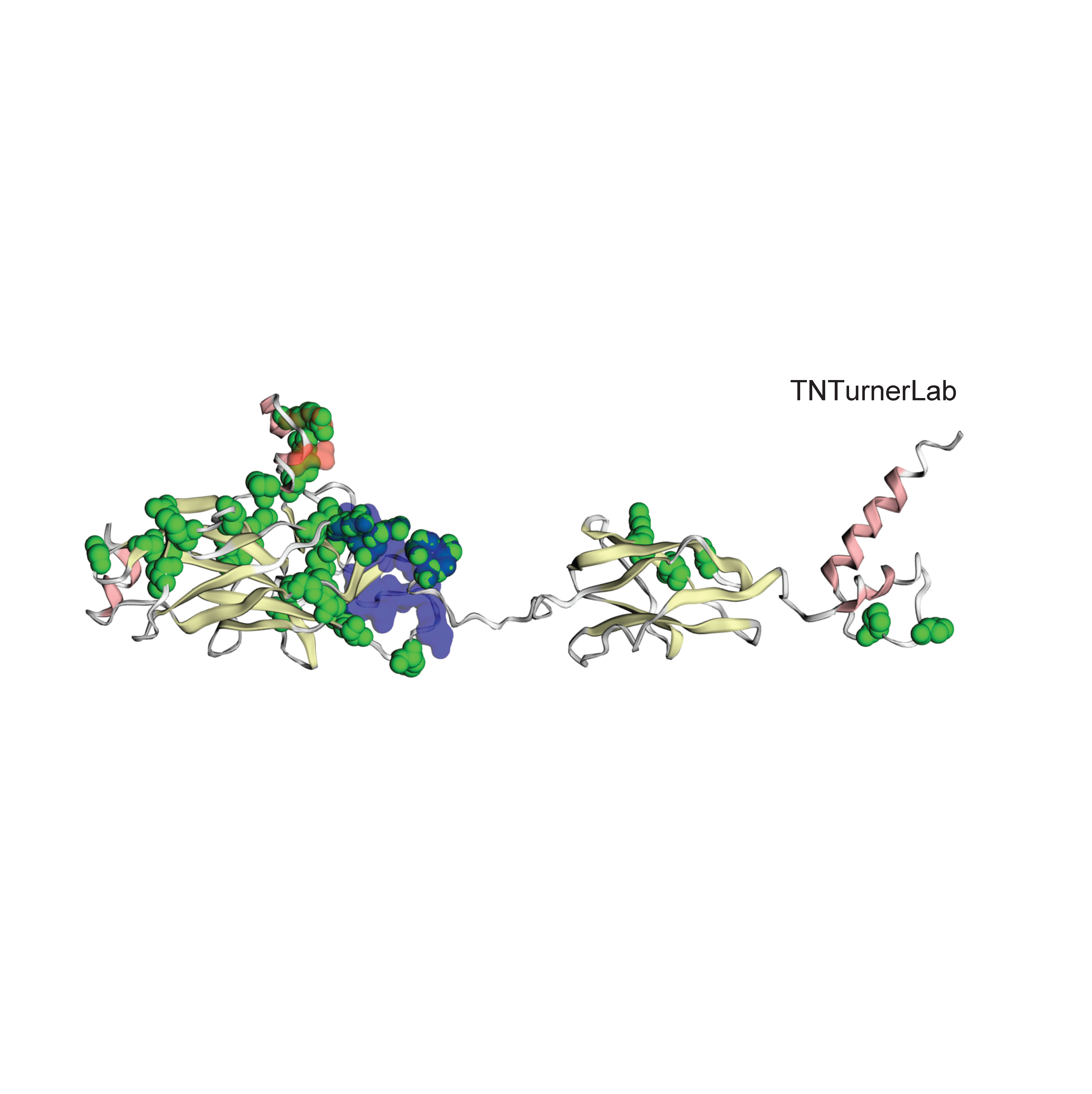
EBF3 is enriched for protein-coding de novo variants in neurodevelopmental disorders
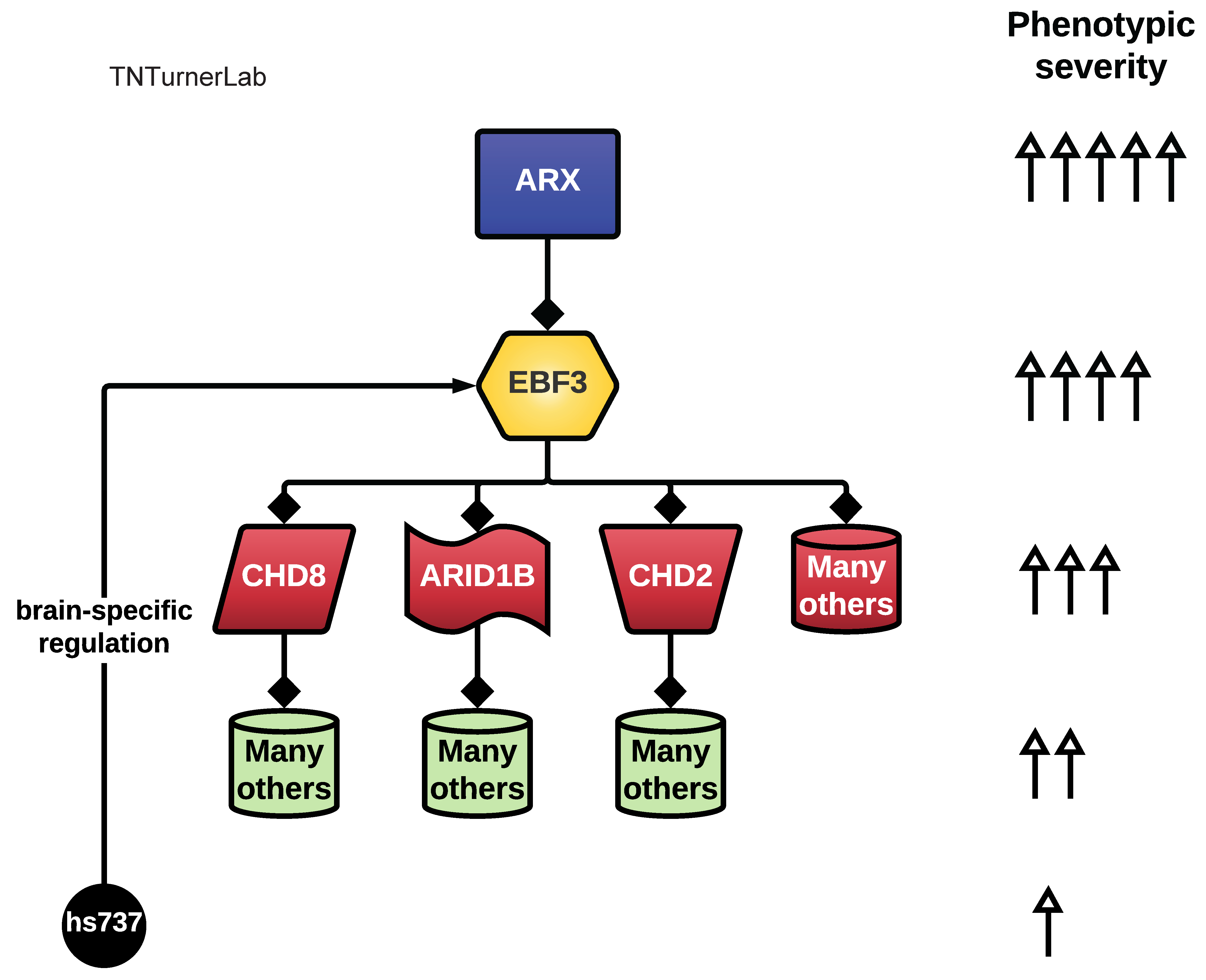
Proposed gene regulatory network for hs737 and EBF3
2. VARIANTS MISSED DUE TO GENOMIC TECHNOLOGY:
In 2001, the first draft of the human genome was announced and within the following few years the genome was “complete.” In the time since then, the field of genomics has consistently developed newer technologies to make advances in surveying human variation genome-wide. First, chromosome microarray technologies allowed checking pre-selected specific variant sites and through clever bioinformatic approaches also enabled discovery of large dosage abnormalities (also referred to as copy number variants). Second, whole-exome sequencing (WES) allowed capture-based assessment of all the protein-coding regions of the genome. By focusing on these regions, researchers have gained insight into high-impact variants underlying a subset of phenotypes. Third, whole-genome sequencing (WGS) using short-read sequencing technologies is the current standard for assessing an individual’s genomic variation and gives access to “all” regions of the genome. However, complex structural variants, likely of large-effect size, are challenging to assess with this approach. Finally, the newest technologies are mostly long-read based and they are allowing access to the full suite of genomic variation including complex structural variants (e.g., translocations, inversions, repeat expansions). We are applying highly-accurate long-read sequencing in neurodevelopmental disorders.
In one of our publications, we studied a family with autism that had no genomic answers from previous technologies. Through long-read sequencing we identified relevant missense variation in KCNC2
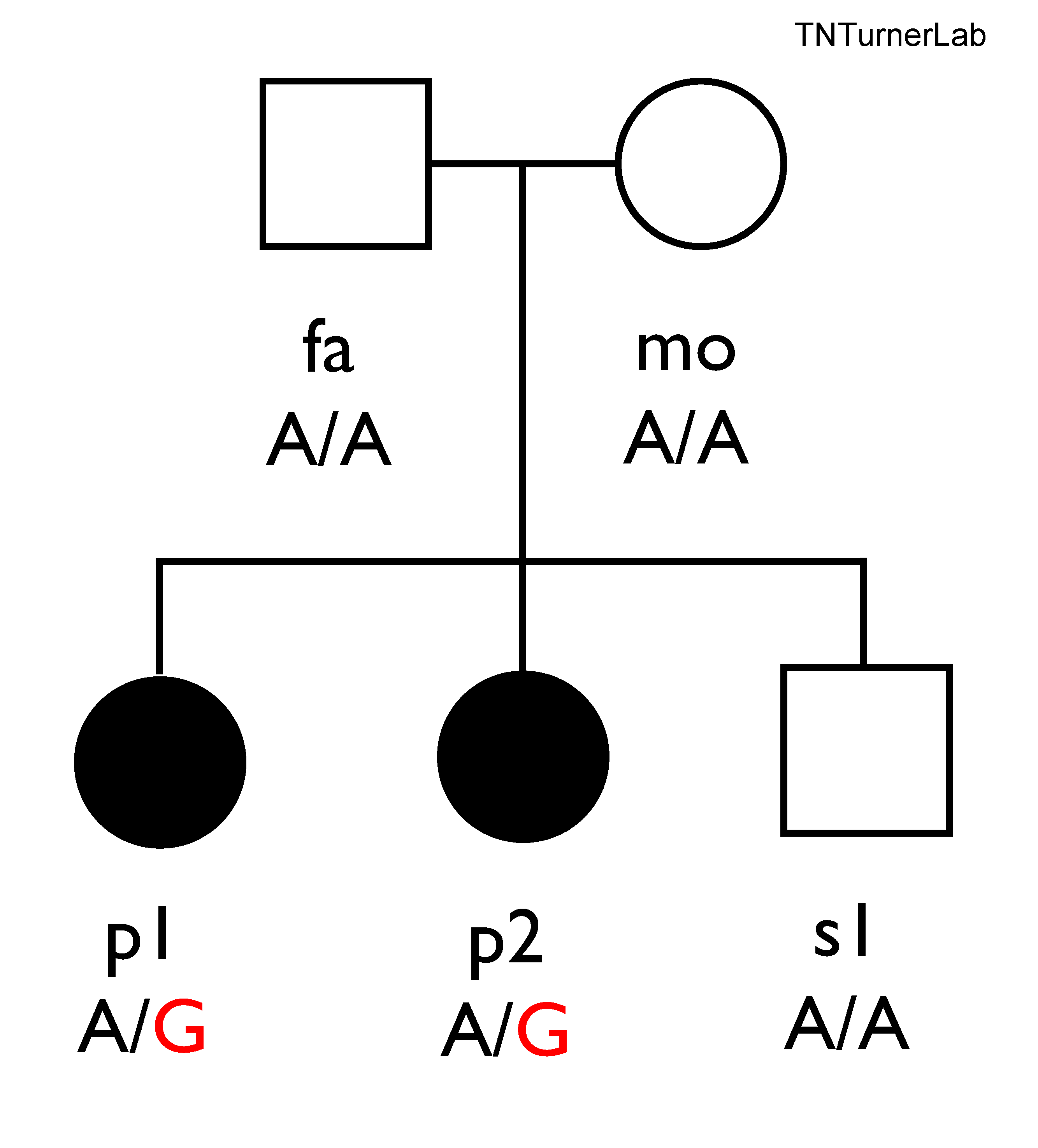
Family we studied using highly-accurate long-read sequencing
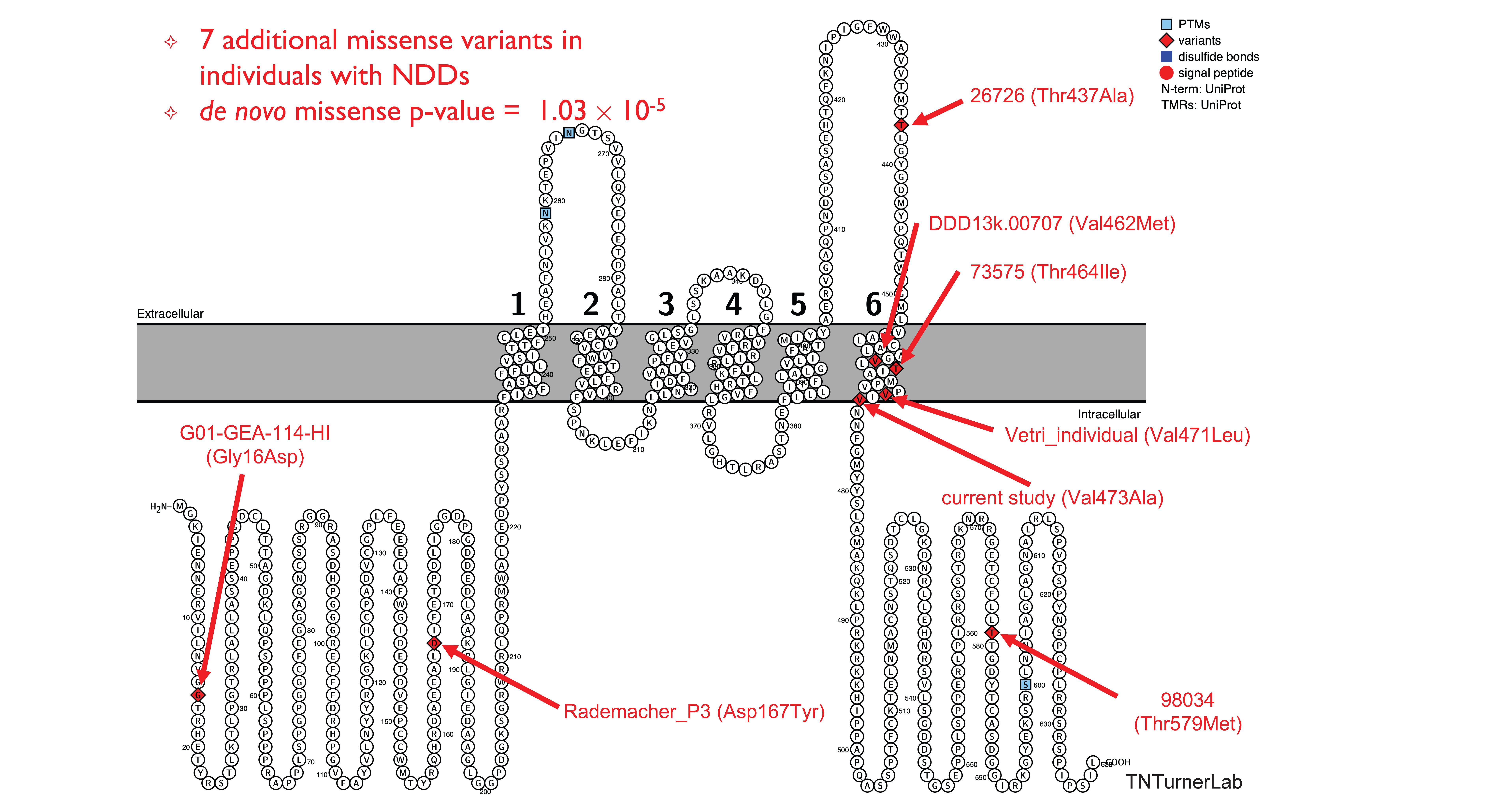
Missense variation in KCNC2 in individuals with NDDs
In another publication, we utilized long-read sequencing to resolve complex variation in an individual with 9p Minus Syndrome. We are currently assessing other individuals with 9p Minus Syndrome using long-read sequencing to gain further insights into this syndrome.
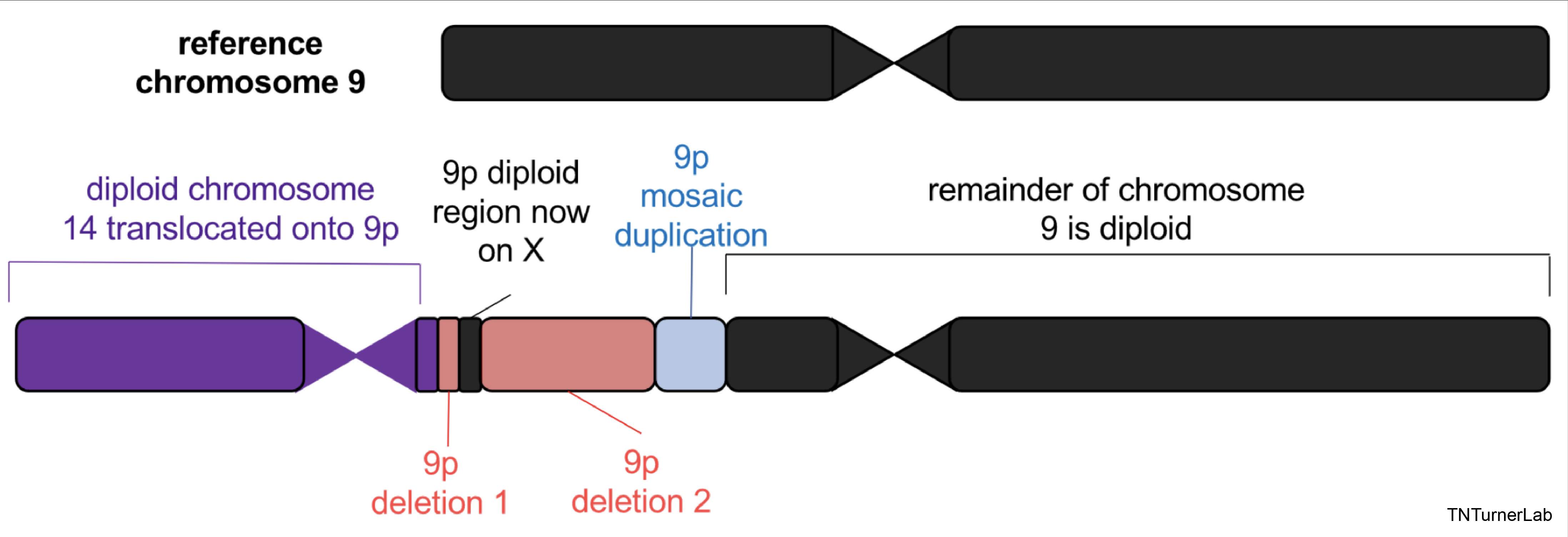
Complex 9p Variation
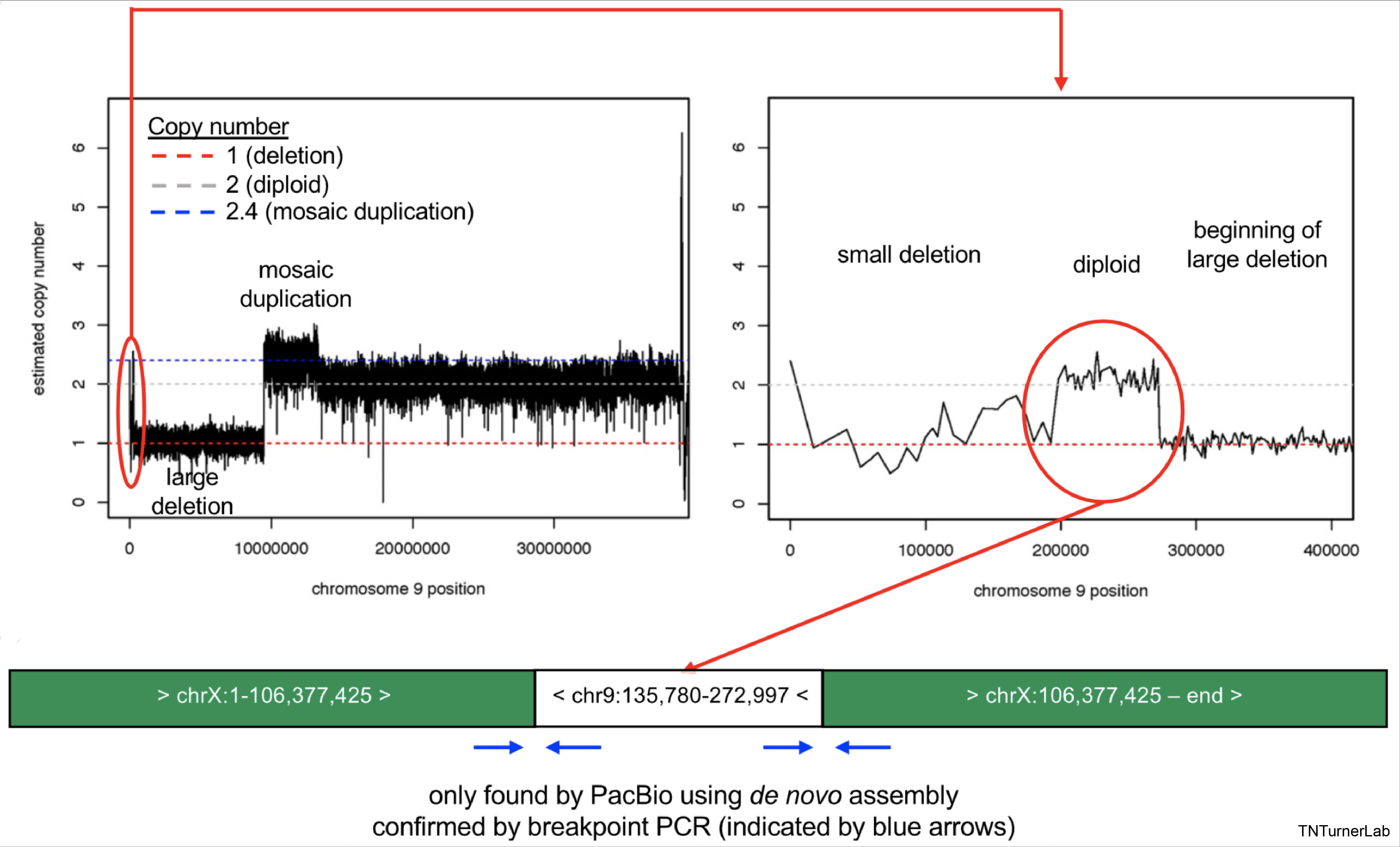
Complex 9p Variation
3. SPEED OF THE CURRENT GENOMIC WORKFLOW:
One key aspect of Precision Genomics is the ability to quickly process genomic information. There are several ways to improve the computational speed including writing more optimized code, parallelizing on more CPU, and utilizing GPUs. We are working on utilizing these different strategies.
In one of our publications, we improved the speed of calling de novo variants ~100x. In this paper, we collaborated with NVIDIA to implement Parabricks as part of our workflow called Hare And Tortoise (HAT). This approach yields highly accurate de novo variant calls in a rapid manner.
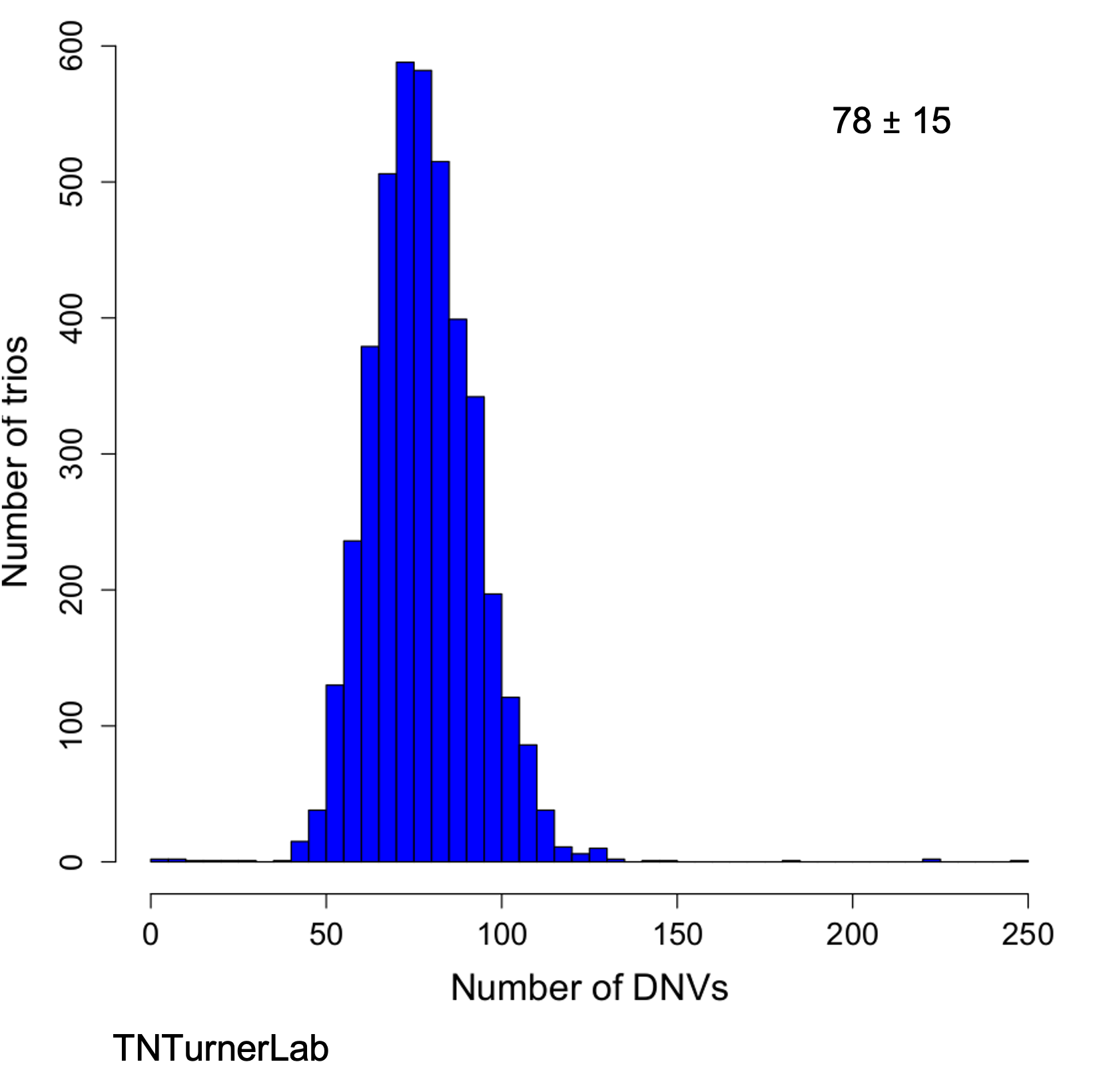
DNVs called with HAT
4. COMBINATION OF MULTI-HIT RARE AND/OR COMMON VARIANTS:
Watch this space. We are working on it.
5. GENE X ENVIRONMENT:
Watch this space. We are working on it.